SEBASTIAN KAULITZKI / TUDOMÁNYOS FOTÓKÖNYVTÁR / Getty Images
A II típusú mukopoliszacharidózis (MPS II), más néven Hunter-szindróma, örökletes rendellenesség, amely az iduronát-2-szulfatáz (I2S) enzim hiányát okozza.Az I2S részt vesz a mukopoliszacharidoknak nevezett komplex szénhidrátok lebontásában. Elegendő I2S hiányában a részben lebontott mukopoliszacharidok felhalmozódnak a test szerveiben és szöveteiben, és mérgezővé válnak.
A Hunter-szindróma egy X-hez kapcsolódó rendellenesség, vagyis az X kromoszómán egy anyától a gyermekeihez terjed. A Hunter-szindróma öröklődése miatt a betegség gyakoribb a férfiaknál - bár ritkán a nők is örökölhetik ezt az állapotot. A Hunter-szindróma bármely etnikai csoportban előfordulhat. Kissé magasabb előfordulási gyakoriságot figyeltek meg az Izraelben élő zsidók körében. Az állapot 100 000-ből 1-ben 150 000 férfiból 1-ben fordul elő.
A Hunter-szindrómának két típusa van - korai és késői.
Korán megjelenő MPS II
A Hunter-szindróma súlyos formáját, a korai megjelenést általában 18-36 hónapos gyermekeknél diagnosztizálják. A várható élettartam ebben a formában változhat, egyes gyermekek életük második és harmadik évtizedéig élnek. A korai betegség tünetei lehetnek:
- durva arcvonások és alacsony termet
- megnagyobbodott máj és lép
- progresszív és mély mentális retardáció
- elefántcsont színű bőrelváltozások a felső kar és a comb felső és hátsó oldalán
- csontvázelváltozások, ízületi merevség, rövid nyak, széles mellkas és túl nagy fej
- progresszív süketség
- atípusos retinitis pigmentosa és látásromlás
Ezek a tünetek hasonlóak a Hurler-szindrómához. A Hurler-szindróma tünetei azonban gyorsabban fejlődnek és rosszabbak, mint a korán megjelenő Hunter-szindróma tünetei.
Késői MPS II
Ez a típusú Hunter-szindróma sokkal enyhébb, mint a korai kialakulás, és csak felnőttkorban diagnosztizálható. A késői betegségben szenvedő személyek várható élettartama sokkal hosszabb, és a 70-es éveikben is élhetnek. Fizikai tulajdonságaik hasonlóak a súlyos MPS II-vel rendelkezőkhöz; az MPS II késői verzióját használók azonban általában normális intelligenciájúak, és hiányoznak a súlyosabb típusú súlyos csontvázproblémák.
Diagnózis
Súlyos Hunter-szindróma esetén a gyermek megjelenése más tünetekkel, például a máj és a lép megnagyobbodásával és az elefántcsontszínű bőrelváltozásokkal (amelyet a szindróma jelzőjeként tartanak számon) arra utalhat, hogy a gyermek mukopoliszacharidózisban szenved. Az enyhe Hunter-szindrómát sokkal nehezebb azonosítani, és csak akkor ismerhető fel, ha Hunter-szindrómás gyermek anyai rokonait vizsgáljuk.
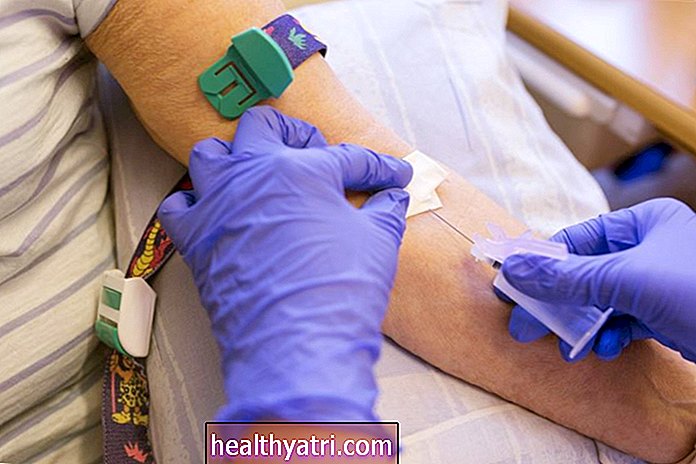
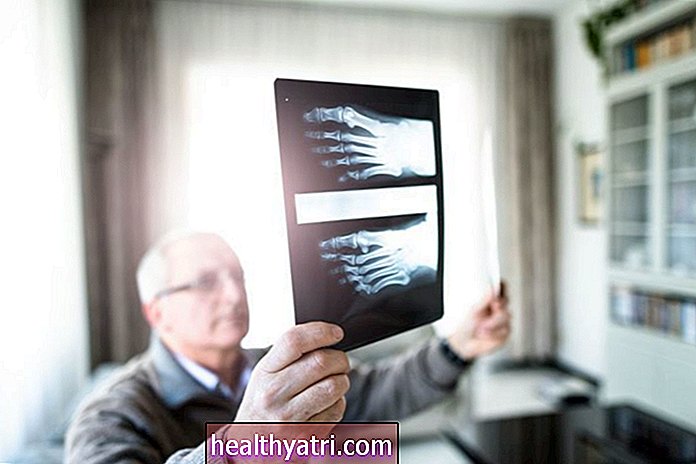
Bármelyik típusban a diagnózis megerősíthető az I2S hiányának vérvizsgálatával. Egy enzimvizsgálat vagy az iduronát-szulfatáz gén változásainak genetikai vizsgálata szintén diagnosztizálhatja az állapotot. A vizeletben mukopoliszacharidok is jelen lehetnek. A röntgensugarak felfedhetik a Hunter-szindrómára jellemző csontváltozásokat.
Az MPS II kezelése
Jelenleg a Hunter-szindrómára nincs gyógymód. Az orvosi ellátás az MPS II tüneteinek enyhítésére irányul. Az Elaprase (idursulfáz) kezelés helyettesíti az I2S-t a szervezetben, és segít csökkenteni a tüneteket és a fájdalmat. A légutak elzáródhatnak, ezért fontos a jó légzésgondozás és monitorozás. Fontos a fizikoterápia és a napi testmozgás. Sok szakember vesz részt a Hunter-szindrómás egyének gondozásában. A genetikai tanácsadó tanácsot adhat családjának és rokonainak a szindróma átadásának kockázataival kapcsolatban.







.jpg)